Generalitate
Amiloidoza este termenul folosit pentru a defini un grup de boli caracterizate prin acumularea, adesea în zona extracelulară, a unui material proteic fibrilar, definit ca amiloid.Fibrilele amiloide insolubile formează depozite deosebit de stabile în numeroase organe.

Alte fotografii Amiloidoză - Galeria 2
Simptomele și severitatea bolii depind de organul afectat predominant de acumularea de amiloid și de tipul de amiloidoză. Cu toate acestea, majoritatea cazurilor sunt sistemice. Cu alte cuvinte, depozitele fibrilare sunt răspândite și pot afecta funcționalitatea multor țesuturi și organe ale corpului . Diagnosticul este definit prin biopsie, prin examinarea unui mic eșantion de țesut la microscop. Factorii etiologici potențiali variază în funcție de varianta amiloidozei. Tratamentele disponibile pot ajuta la gestionarea simptomelor și la limitarea producției de amiloid.
Caracteristicile depozitelor de amiloid
Amiloidoza derivă din tulburări ale structurii secundare a proteinelor (cu configurație de foaie pliată β). În condiții normale, de fapt, proteinele sunt sintetizate într-un șir liniar de aminoacizi, care, atunci când sunt pliate, iau o conformație spațială specifică (plierea proteinelor). Datorită structurii sale, deci plierii corecte a proteinelor, proteina este capabilă să îndeplinească funcțiile fiziologice de care este responsabilă. Proteinele amiloide derivă dintr-un precursor procesat incorect de celule (datorită „plierii greșite”) Proteinele care formează fibrile sunt diversificate în funcție de mărime, secvența de aminoacizi și structura nativă, dar devin agregate insolubile care sunt similare ca structură și proprietăți. Precursorii fibrilelor sunt reprezentați de molecule primare (exemplu: lanț ușor de imunoglobuline, β2-microglobulină, apolipoproteină A1 etc.) sau de la produse care reflectă o alterare în s echența aminoacizilor. Structura secundară aberantă predispune la formarea fibrilelor, care pot fi depuse local în țesuturi și organe și pot duce la afectarea funcției lor fiziologice normale. Au fost identificați mai mult de 20 de precursori de proteine care pot lua o conformație amiloidă, care este de ce există multe tipuri diferite de amiloidoză.
Pe baza localizării depozitelor de amiloid, boala poate fi împărțită în:
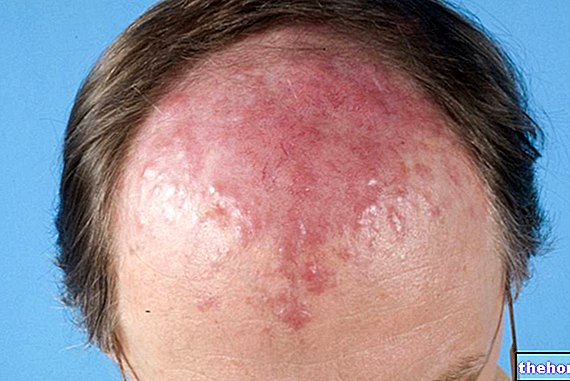
- Formă localizată: limitată la un anumit organ sau țesut (inimă, rinichi, tract gastrointestinal, sistem nervos și derm) și este de obicei mai puțin severă decât formele sistemice (difuze). De exemplu, amiloidoza poate afecta doar pielea, provocând decolorare și / sau mâncărime. Un anumit tip de proteină amiloidă a fost găsit și în creierul pacienților cu boala Alzheimer. Amiloidoza localizată este tipică senescenței și a pacienților afectați. Din diabetul de tip 2 (unde proteinele se acumulează în pancreas).
- Forma sistemică: depozitele de amiloid sunt prezente în diferite organe și recunosc în general originea neoplazică, inflamatorie, genetică sau iatrogenă. Amiloidoza sistemică este adesea foarte gravă: dăunează frecvent inimii, rinichilor, intestinelor și nervilor, provocând insuficiență progresivă d "organ.
Clasificare
Există multe forme de amiloidoză, clasificate în funcție de natura proteinelor care alcătuiesc depozitele fibrilare.
Cele mai frecvente variante sunt:
- Amiloidoză primară (numită și amiloidoză cu lanț ușor, AL);
- Amiloidoză secundară (numită și amiloidoză dobândită, AA);
- Amiloidoză ereditară;
- Amiloidoza asociată îmbătrânirii (sau amiloidoza sistemică senilă).