Ingrediente active: Filgrastim
Nivestim 12 MU / 0,2 ml soluție injectabilă / perfuzabilă
Nivestim 30 MU / 0,5 ml soluție injectabilă / perfuzabilă
Nivestim 48 MU / 0,5 ml soluție injectabilă / perfuzabilă
De ce se utilizează Nivestim? Pentru ce este?
Ce este Nivestim
Nivestim conține substanța activă filgrastim care aparține unui grup de proteine numite citokine și este foarte asemănător cu o proteină naturală (factor de stimulare a coloniei de granulocite [G-CSF]) produsă de corpul uman. Filgrastim stimulează măduva osoasă (țesutul care produce celule sanguine noi) pentru a produce mai multe celule sanguine, în special unele tipuri de celule albe din sânge. Celulele albe din sânge sunt importante deoarece ajută organismul să lupte împotriva infecțiilor.
Pentru ce se utilizează Nivestim
Medicul dumneavoastră v-a prescris Nivestim pentru a vă ajuta corpul să producă mai multe celule albe din sânge. Medicul dumneavoastră vă va explica de ce vi s-a prescris Nivestim. Nivestim este util în mai multe condiții clinice diferite, cum ar fi:
- chimioterapie
- transplant de măduvă osoasă,
- neutropenie cronică severă (neutropenia este un număr scăzut anormal al unui anumit tip de celule albe din sânge, cunoscute și sub numele de neutrofile),
- neutropenie la pacienții cu infecție cu HIV,
- mobilizarea celulelor stem din sângele periferic.
Contraindicații Când nu trebuie utilizat Nivestim
Nu utilizați Nivestim
- dacă sunteți alergic la filgrastim sau la oricare dintre celelalte componente ale acestui medicament.
Precauții pentru utilizare Ce trebuie să știți înainte de a lua Nivestim
Înainte să luați Nivestim, adresați-vă medicului dumneavoastră, farmacistului sau asistentei medicale: - dacă aveți orice altă boală (mai ales dacă credeți că aveți o „infecție),
- dacă aveți tuse, febră și dificultăți de respirație. Acestea ar putea fi secundare problemelor pulmonare (vezi și secțiunea 4 „EVENIMENTE ADVERSE POSIBILE”),
- dacă aveți anemie falciformă (o afecțiune sanguină moștenită care afectează globulele roșii),
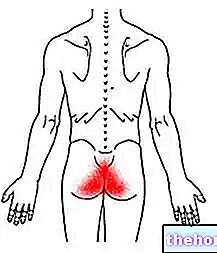
- dacă aveți dureri abdominale în partea stângă sus sau dacă aveți dureri de umăr. Ar putea fi o consecință a unei boli a splinei (vezi secțiunea 4 „EVENIMENTE ADVERSE POSIBILE”),
- dacă suferiți de tulburări sanguine specifice (de exemplu, sindromul Kostmann, sindromul mielodisplazic, diferite tipuri de leucemie),
- dacă suferiți de osteoporoză. Medicul dumneavoastră vă poate verifica în mod regulat densitatea oaselor.
Dacă trebuie să faceți o scanare osoasă, vă rugăm să spuneți medicului dumneavoastră sau asistentei medicale că sunteți tratat cu Nivestim.
Spuneți imediat medicului dumneavoastră sau asistentei medicale dacă prezentați semne bruște de alergie, cum ar fi erupții cutanate, mâncărime sau urticarie pe piele, umflarea feței, buzelor, limbii sau a altor părți ale corpului, dificultăți de respirație, respirație șuierătoare sau dificultăți de respirație în timpul tratamentului. cu Nivestim deoarece acestea pot fi semne ale unei reacții alergice severe.
În timp ce sunteți tratat cu Nivestim, este posibil să aveți nevoie de teste periodice de sânge pentru a verifica numărul de neutrofile și alte celule albe din sânge. Cu aceste date, medicul determină eficacitatea tratamentului și dacă acesta ar trebui să continue.
Pierderea răspunsului la filgrastim
Dacă ați scăzut răspunsul sau nu ați menținut răspunsul la tratamentul cu filgrastim, medicul dumneavoastră va investiga motivele, inclusiv posibilitatea de a fi dezvoltat anticorpi care neutralizează activitatea filgrastimului.
Interacțiuni Ce medicamente sau alimente pot modifica efectul Nivestim
Nu trebuie să fiți tratat cu Nivestim în 24 de ore înainte și 24 de ore după tratamentul cu chimioterapie.
Spuneți medicului dumneavoastră sau farmacistului dacă luați, ați luat recent sau s-ar putea să luați orice alte medicamente.
Avertismente Este important să știm că:
Sarcina și alăptarea
Adresați-vă medicului dumneavoastră sau farmacistului pentru recomandări înainte de a lua orice medicament.
Filgrastim nu a fost studiat la femeile gravide. Este important să spuneți medicului dumneavoastră dacă sunteți gravidă, dacă credeți că ați putea fi gravidă sau dacă intenționați să rămâneți gravidă, deoarece medicul dumneavoastră poate decide că nu puteți utiliza acest medicament. Filgrastim poate avea efecte adverse asupra capacitatea de a rămâne gravidă sau de a duce la încheierea unei sarcini.
Nu se știe dacă filgrastimul trece în laptele matern. Prin urmare, medicul dumneavoastră poate decide că nu puteți utiliza acest medicament dacă alăptați.
Conducerea vehiculelor și utilizarea utilajelor
Filgrastim are efecte modeste asupra capacității de a conduce vehicule și de a folosi utilaje. Dacă vă simțiți obosit, trebuie să aveți grijă când conduceți vehicule sau folosiți utilaje.
Nivestim conține sorbitol
Acest medicament conține sorbitol (E420). Dacă medicul dumneavoastră v-a spus că aveți intoleranță la unele zaharuri (fructoză), contactați medicul înainte de a lua acest medicament. Acest medicament conține, de asemenea, sodiu în mai puțin de 1 mmol (23 mg) pe doză și, prin urmare, este în esență „fără sodiu”.
Doză, metodă și timp de administrare Cum se utilizează Nivestim: Doze
Utilizați întotdeauna acest medicament exact așa cum v-a spus medicul dumneavoastră. Adresați-vă medicului dumneavoastră dacă nu sunteți sigur.
Acest medicament se administrează prin injecție, fie prin perfuzie intravenoasă (picurare), fie subcutanat în țesutul situat direct sub piele.
Dacă utilizați acest medicament prin injecție subcutanată, medicul dumneavoastră vă poate considera potrivit să îl injectați singur. Medicul sau asistenta vă vor arăta cum să vă injectați (instrucțiunile despre auto-injectare sunt date la sfârșitul acestui prospect). Nu încercați să vă injectați singur decât dacă ați fost instruit în acest sens. Unele informații de care aveți nevoie sunt raportate în partea de jos din acest prospect, însă, pentru tratamentul adecvat al bolii dumneavoastră, este necesară o cooperare atentă și constantă cu medicul dumneavoastră.
Cantitatea de Nivestim de care aveți nevoie depinde de boala pentru care luați Nivestim și de greutatea corporală.
Nivestim și neutropenie asociată chimioterapiei
Doza uzuală la adulți și copii este de 0,5 milioane de unități (5 micrograme) pe kilogram de greutate corporală pe zi. De exemplu, dacă cântăriți 60 kg, doza zilnică este de 30 de milioane de unități (300 micrograme). Tratamentul poate dura până la 14 zile. Pentru unele boli, poate fi necesar un tratament prelungit de până la aproximativ o lună.
Nivestim și transplant de măduvă osoasă
Doza inițială uzuală este de 1 milion de unități (10 micrograme) pe kilogram de greutate corporală pe zi prin perfuzie. De exemplu, dacă cântăriți 60 kg, doza zilnică este de 60 de milioane de unități (600 micrograme). Veți primi, în general, prima doză cel puțin 24 de ore după chimioterapie, dar în termen de 24 de ore de la transplantul de măduvă osoasă. Medicul va efectua apoi analize de sânge pentru a verifica efectul tratamentului și pentru a stabili durata acestuia.
Nivestim și neutropenie cronică severă
Doza inițială uzuală este cuprinsă între 0,5 milioane (5 micrograme) și 1,2 milioane (12 micrograme) de unități pe kilogram de greutate corporală pe zi, sub formă de doză unică sau divizată. Medicul dumneavoastră va efectua apoi analize de sânge pentru a verifica efectul tratamentului și pentru a determina doza cea mai potrivită pentru dvs. În caz de neutropenie, este necesar un tratament prelungit cu Nivestim.
Nivestim și neutropenie la pacienții cu infecție cu HIV
Doza inițială uzuală este între 0,1 (1 microgramă) și 0,4 milioane de unități (4 micrograme) pe kilogram de greutate corporală pe zi. Medicul dumneavoastră va efectua teste de sânge la intervale regulate pentru a verifica efectul tratamentului. Odată ce numărul de globule albe a revenit la normal, frecvența dozelor poate fi redusă la mai puțin de o dată pe zi. Menținerea numărului normal de globule albe poate necesită tratament prelungit cu Nivestim.
Nivestim și transplant de celule stem din sângele periferic
Dacă donați celule stem pentru dvs., doza uzuală este de 0,5 milioane (5 micrograme) la 1 milion de unități (10 micrograme) per kilogram de greutate corporală pe zi. Tratamentul cu Nivestim durează până la 2 săptămâni. Medicul va efectua analize de sânge pentru a determina cel mai bun moment pentru colectarea celulelor stem.
Dacă donați celule stem pentru o altă persoană, doza uzuală este de 1 milion de unități pe kilogram de greutate corporală pe zi. Tratamentul cu Nivestim va dura 4 până la 5 zile.
Dacă uitați să utilizați Nivestim
Dacă ați uitat să vă injectați o doză, contactați medicul sau farmacistul și întrebați-i când să vă injecteze următoarea doză. Nu utilizați o doză dublă pentru a compensa injecția uitată.
Cum se termină tratamentul Nivestim
Medicul dumneavoastră vă va spune când să încetați să utilizați Nivestim. Este destul de normal să aveți mai multe cure de tratament cu Nivestim.
Dacă aveți orice întrebări suplimentare cu privire la utilizarea medicamentului, adresați-vă medicului dumneavoastră sau farmacistului.
Supradozaj Ce trebuie făcut dacă ați luat prea mult Nivestim
Dacă utilizați mai mult Nivestim decât trebuie, contactați medicul sau farmacistul cât mai curând posibil.
Efecte secundare Care sunt efectele secundare ale Nivestim
Ca toate medicamentele, acest medicament poate provoca reacții adverse, deși nu apar la toate persoanele.
Au fost raportate reacții alergice la filgrastim, inclusiv erupții cutanate, zone cu mâncărime crescută și anafilaxie (slăbiciune, scăderea tensiunii arteriale, dificultăți de respirație și înghițire și umflarea feței). Dacă credeți că aveți aceste tipuri de reacții, opriți administrarea de injecții Nivestim și solicitați imediat asistență medicală.
Au fost raportate dimensiuni crescute ale splinei și cazuri foarte rare de rupere a splinei. Unele cazuri de rupere a splinei au fost fatale. Este important să vă adresați imediat medicului dumneavoastră dacă aveți dureri la nivelul abdomenului superior stâng sau al omoplatului, deoarece acest lucru poate fi un semn al problemelor cu splina.
Spuneți imediat medicului dumneavoastră dacă aveți unul sau mai multe dintre următoarele evenimente adverse în timpul tratamentului:
- umflături sau umflături, care pot fi legate de scăderea urinării, dificultăți de respirație, balonare și senzație de plin și o senzație generală de oboseală. Aceste simptome apar de obicei rapid.
Acestea pot fi simptome ale unei boli mai puțin frecvente (pot afecta până la 1 din 100 de persoane) cunoscută sub numele de „Sindrom de scurgere capilară”, care determină scurgerea de sânge în corp din vasele de sânge mici și care necesită asistență medicală urgentă.
De asemenea, este foarte important să spuneți medicului dumneavoastră dacă credeți că aveți o „infecție. Există multe moduri în care se poate manifesta o” infecție. Trebuie să verificați dacă temperatura nu este și nu depășește 37,8 ° C, frisoane sau alte semne de infecție, cum ar fi roșeață a pielii, dureri în gât, diaree, dureri de urechi, dificultăți sau dureri în respirație sau probleme precum tuse și astm. Aceste simptome pot se datorează efectelor adverse pulmonare severe, cum ar fi pneumonia și sindromul de detresă respiratorie la adulți, care pot provoca moartea. Dacă aveți febră sau orice alte simptome, contactați imediat medicul dumneavoastră și mergeți direct la spital.
Dacă aveți anemie falciformă, asigurați-vă că vă informați medicul înainte de a începe tratamentul cu Nivestim. Au fost raportate crize de anemie falciformă la unii pacienți cu această boală tratați cu filgrastim.
Evenimente adverse foarte frecvente (afectează mai mult de 1 din 10 pacienți)
- Senzație sau rău
- Durere în oase și mușchi. Adresați-vă medicului dumneavoastră ce medicament să luați pentru a ameliora acest lucru
- Sângerarea nasului
- Scăderea nivelului de glucoză din sânge care vă poate face să vă simțiți flămând, bolnav, slab, obosit, tremurat sau confuz sau să provocați transpirații, dureri de cap, vedere încețoșată sau creșterea ritmului cardiac
- Creșterea valorilor enzimelor hepatice sau modificări ale analizelor de sânge. Medicul dumneavoastră vă va face analize de sânge pentru a verifica acest lucru
- Creșterea acidului uric care se poate manifesta cu gută
- Dureri în piept
Evenimente adverse frecvente (afectează până la 1 din 10 pacienți)
- Slăbiciune
- Oboseală generalizată
- Durere de cap
- Constipație sau diaree
- Pierderea poftei de mâncare
- Inflamația și ulcerarea gurii și a mucoasei interioare a intestinului
- Tuse
- Durere de gât
- Pierderea parului
- Eczemă
- Ficatul mărit
- Subțierea oaselor
- Durere la locul injectării
- Inflamația vaselor de sânge
- Caderea trombocitelor (celule implicate în coagulare) - cu risc crescut de sângerare sau vânătăi
Evenimente adverse mai puțin frecvente (afectează până la 1 din 100 de pacienți)
- Durere nespecificată
- Prezența sângelui sau a proteinelor în urină
Evenimente adverse rare (afectează până la 1 din 1000 de pacienți)
- Afectarea ficatului cauzată de blocarea venelor mici din ficat (boala veno-ocluzivă)
- O modificare a reglării fluidelor din organism care poate duce la umflături
Evenimente adverse foarte rare (afectează până la 1 din 10000 de pacienți)
- Radiografie anormală cu raze X a plămânilor (infiltrare pulmonară)
- Leziuni violete, ridicate și dureroase la nivelul membrelor și uneori la nivelul feței și gâtului, asociate cu febră (sindromul Sweet)
- Inflamația vaselor de sânge ale pielii (vasculită cutanată) • Agravarea artritei reumatoide deja prezentă
- Modificare neobișnuită a urinei
Frecvență necunoscută (frecvența nu poate fi estimată din datele disponibile)
- Umflături și dureri la nivelul articulațiilor, ca în gută (pseudogută).
Boala grefă versus gazdă (GvHD) poate apărea la pacienții supuși donării de celule stem sau transplant de măduvă osoasă - aceasta este o reacție a celulelor donatoare față de persoana care primește transplantul; semnele și simptomele includ erupții cutanate pe palme sau tălpi și ulcere și răni. în gură, intestine, ficat, piele sau ochi, plămâni, vagin și articulații. Unele cazuri de GvHD au fost fatale.
Evenimentele adverse pe care le-ați putea experimenta dacă sunteți donator de celule stem pentru o altă persoană sunt:
Evenimente adverse foarte frecvente (afectează mai mult de 1 din 10 pacienți)
- Durere de cap
- Durere în oase sau mușchi. Adresați-vă medicului dumneavoastră ce medicament să luați pentru a ameliora acest lucru
- Modificări ale globulelor albe sau ale trombocitelor (medicul dumneavoastră le va verifica cu analize de sânge)
Evenimente adverse frecvente (afectează până la 1 din 10 pacienți)
- Creșterea nivelului unor enzime hepatice (medicul dumneavoastră le va monitoriza)
Evenimente adverse mai puțin frecvente (afectează până la 1 din 100 de pacienți)
- Reacție alergică severă
- Probleme cu splina
- Niveluri crescute de acid uric care pot apărea cu gută
- Agravarea artritei reumatoide
Raportarea efectelor secundare
Dacă manifestați orice reacții adverse, adresați-vă medicului dumneavoastră sau farmacistului sau asistentei medicale.Acestea includ orice reacții adverse posibile care nu sunt enumerate în acest prospect. De asemenea, puteți raporta reacțiile adverse direct prin intermediul sistemului național de raportare enumerat în Anexa V.
Expirare și reținere
Nu lăsați acest medicament la vederea și îndemâna copiilor.
Nu utilizați acest medicament după data de expirare înscrisă pe cutie și pe eticheta seringii după abrevierea EXP / EXP. Data de expirare se referă la ultima zi a lunii.
A se păstra și transporta la frigider (2 ° C - 8 ° C). Nu înghețați. Păstrați seringa preumplută în ambalajul original pentru a proteja medicamentul de lumină.
Seringa poate fi ținută afară din frigider și lăsată la temperatura camerei pentru o singură perioadă de până la 7 zile (cu toate acestea, nu peste 25 ° C).
Nu utilizați Nivestim dacă observați că este tulbure sau că există particule în el.
Nu aruncați niciun medicament pe calea apei uzate sau a deșeurilor menajere. Întrebați farmacistul cum să aruncați medicamentele pe care nu le mai utilizați. Acest lucru va ajuta la protejarea mediului.
Alte informații
Ce conține Nivestim
- Ingredientul activ este filgrastim. Fiecare mililitru conține 60 de milioane de unități [UM] (600 micrograme) sau 96 de milioane de unități [UM] (960 micrograme) de filgrastim.
- Nivestim 12 MU / 0,2 ml soluție injectabilă / perfuzabilă: fiecare seringă preumplută conține 12 milioane de unități (UM), 120 micrograme de filgrastim în 0,2 ml (corespunzând la 0,6 mg / ml).
- Nivestim 30 MU / 0,5 mL soluție injectabilă / perfuzabilă: Fiecare seringă preumplută conține 30 de milioane de unități (MU), 300 micrograme de filgrastim în 0,5 mL (corespunzător la 0,6 mg / mL).
- Nivestim 48 MU / 0,5 mL soluție injectabilă / perfuzabilă: Fiecare seringă preumplută conține 48 de milioane de unități (MU), 480 micrograme de filgrastim în 0,5 mL (corespunzător la 0,96 mg / mL).
- Celelalte componente sunt acid acetic (glacial), hidroxid de sodiu, sorbitol E420, polisorbat 80 și apă pentru preparate injectabile.
Cum arată Nivestim și conținutul ambalajului
Nivestim este o soluție limpede, incoloră, pentru injecție / perfuzie, într-o seringă preumplută cu un ac de injecție protector (din oțel inoxidabil). Pot fi 1, 5, 8 sau 10 seringi în fiecare ambalaj.
Instrucțiuni pentru auto-injectare pentru pacienți
Această secțiune conține informații despre modul de injectare a Nivestim. Este important să nu încercați să vă injectați singur medicamentul până când nu ați fost instruit special de medicul sau asistenta medicală.
De asemenea, este important să aruncați seringa într-un recipient rezistent la înțepături. Dacă aveți întrebări sau nelămuriri cu privire la auto-injectare, adresați-vă medicului sau asistentei.
Cum mă injectez?
În mod normal, Nivestim se administrează de două ori pe zi prin injecție, în țesuturile situate sub piele, cunoscută și sub numele de injecție subcutanată.
Învățarea procedurii de autoinjecție înseamnă evitarea așteptării la domiciliu a sosirii asistentei, cu atât mai puțin a fi nevoie să mergi la spital sau clinică în fiecare zi pentru a primi injecția.
Va trebui să vă injectați la aceeași oră în fiecare zi. Cele mai potrivite locuri pentru injectare sunt:
- partea din față a coapsei,
- abdomenul, cu excepția zonei din jurul buricului.
Cel mai bine este să schimbați întotdeauna locul injectării în fiecare zi pentru a evita durerea într-o anumită zonă.
Echipament necesar administrării
Următoarele echipamente sunt necesare atunci când se efectuează o "auto-injecție subcutanată:"
- O nouă seringă preumplută Nivestim.
- Un recipient pentru obiecte ascuțite (rezistent la perforarea acului) pentru a elimina în siguranță seringile uzate.
- Șervețele antiseptice (dacă este recomandat de medicul sau asistenta medicală).
Cum pot face autoinjecția subcutanată Nivestim?
- Încercați să vă injectați la aproximativ aceeași oră în fiecare zi.
- Scoateți seringa Nivestim din frigider și lăsați-o să atingă temperatura camerei (aproximativ 25 ° C). Aceasta va dura 15-30 de minute. Verificați data de pe ambalaj pentru a vă asigura că medicamentul nu a expirat. Aveți recipientul pentru colectare a deșeurilor ascuțite din apropiere.
- Căutați un loc de lucru bine luminat pentru injectare și verificați doza prescrisă.
- Spălați-vă bine mâinile cu apă și săpun.
- Scoateți seringa din blister și verificați dacă soluția pe care o conține este limpede, incoloră și practic lipsită de particule vizibile. Nu utilizați seringa Nivestim dacă lichidul conține particule plutitoare sau dacă o parte din lichid s-a scurs din seringă.
- Țineți seringa cu acul îndreptat în sus. Scoateți capacul de protecție din ac Seringa este acum gata de utilizare. Poate exista o mică bulă de aer în seringă. Nu trebuie să îndepărtați bulele de aer înainte de injectare. Injectarea soluției în prezența unei bule de aer nu este periculoasă.
- Decideți unde să injectați Nivestim - găsiți un loc pe partea din față a abdomenului sau pe partea din față a coapselor. Selectați de fiecare dată un loc de injectare diferit. Nu alegeți o zonă dureroasă, roșie, învinețită sau cu cicatrici.Dacă asistenta sau medicul vă recomandă, curățați suprafața pielii cu un dezinfectant.
- Luați o „cută largă a pielii între degetul mare și arătător, având grijă să nu atingeți zona pe care tocmai ați curățat-o”.
- Cu cealaltă mână, introduceți acul sub piele la un unghi de aproximativ 45 °.
- Trageți ușor pistonul pentru a verifica dacă nu intră sânge în seringă. Dacă vedeți sânge în seringă, scoateți acul și introduceți-l în altă parte. Împingeți încet pistonul până când întregul conținut al seringii a fost golit.
- După injectarea soluției, scoateți acul de pe piele.
- Urmând instrucțiunile de mai jos pentru dispozitivul activ sau pasiv de protecție a acului, asigurați-vă că dispozitivul acoperă acul.
- Întoarceți seringa în recipientul pentru obiecte ascuțite. Nu încercați să înlocuiți capacul de protecție.
- Nu lăsați seringile uzate la îndemâna și vederea copiilor.
- NU aruncați NICIODATĂ seringi în recipientul pentru deșeuri menajere.
Tine minte
Majoritatea oamenilor pot afla despre auto-injectarea subcutanată, dar dacă aveți o mulțime de dificultăți, nu vă fie teamă să cereți ajutor medicului sau asistentei medicale sau sfaturi.
Utilizarea Active Ultrasafe Needle Guard pentru Nivestim 12 MU / 0,2 ml soluție injectabilă / perfuzabilă
Seringa preumplută este echipată cu un dispozitiv de siguranță al acului, UltraSafe Needle Guard, care protejează împotriva lipirii accidentale a acului. Când manipulați seringa preumplută, țineți-vă mâinile în spatele acului.
- Efectuați injecția conform tehnicii descrise mai sus.
- După ce ați terminat injecția, glisați protecția acului înainte până când acul este complet acoperit (dispozitivul se fixează în poziție).
Utilizarea Ultrasafe Passive Needle Guard pentru Nivestim 30 MU / 0,5 ml soluție injectabilă / perfuzabilă și Nivestim 48 MU / 0,5 ml soluție injectabilă / perfuzabilă
Seringa preumplută este echipată cu un dispozitiv de protecție împotriva acului, UltraSafe Needle Guard, care protejează împotriva lipirii accidentale a acului. Când manipulați seringa preumplută, țineți-vă mâinile în spatele acului.
- Efectuați injecția conform tehnicii descrise mai sus.
- În timp ce țineți seringa cu degetele sprijinite pe marginea sa de sprijin, aplicați presiunea asupra pistonului până când injecția întregii doze este completă. Sistemul pasiv de protecție a acului NU se va activa dacă nu a fost administrată doza FULL.
- Scoateți acul din piele, apoi eliberați pistonul și seringa se va deplasa înainte până când scutul a acoperit acul și se fixează în poziție.
INFORMAȚIILE URMĂTOARE sunt destinate numai medicilor sau profesioniștilor din domeniul sănătății
Nivestim nu conține conservanți. Datorită riscului de contaminare bacteriană, seringile Nivestim sunt de unică folosință.
Expunerea accidentală la temperaturi de îngheț de până la 24 de ore nu are niciun efect negativ asupra stabilității Nivestim. Seringa preumplută congelată poate fi decongelată și returnată la frigider pentru utilizare ulterioară. Dacă expunerea la temperaturi scăzute este mai mare de 24 de ore sau dacă este înghețată de mai multe ori, Nivestim NU mai trebuie utilizat.
Nivestim nu trebuie diluat în soluție de clorură de sodiu. Acest medicament nu trebuie amestecat cu alte medicamente, cu excepția celor descrise mai jos. Dacă nu este diluat așa cum este descris mai jos, filgrastimul diluat se poate absorbi în sticlă și materiale plastice.
Dacă este necesar, Nivestim poate fi diluat în soluție perfuzabilă de glucoză 50 mg / ml (5%). Nu se recomandă niciodată diluarea la concentrații finale <0,2 MU (2 micrograme) per ml. Soluția trebuie inspectată vizual înainte de utilizare. Trebuie utilizate numai soluții clare fără particule vizibile. Pentru pacienții tratați cu filgrastim diluat la concentrații sub 1,5 MU (15 micrograme) per ml, albumina serică umană (HSA) până la o concentrație finală de 2 mg / ml.
Exemplu: într-un volum final de injectat de 20 ml, doze totale de filgrastim sub 30 MU (300 micrograme) trebuie administrate prin adăugarea a 0,2 ml dintr-o soluție de albumină umană de 200 mg / ml (20%). Diluat cu soluție perfuzabilă de glucoză 50 mg / ml (5%), Nivestim este compatibil cu sticla și diverse materiale plastice precum PVC, poliolefină (un copolimer de polipropilenă și polietilenă) și polipropilenă.
După diluare: stabilitatea chimică și fizică în utilizare a soluției diluate pentru perfuzie a fost demonstrată timp de 24 de ore la 2 ° C până la 8 ° C. Din punct de vedere microbiologic, produsul trebuie utilizat imediat. Dacă nu este utilizat imediat, timpii și condițiile de păstrare înainte de utilizare sunt responsabilitatea utilizatorului și în mod normal nu ar depăși 24 de ore la 2 ° C până la 8 ° C, cu excepția cazului în care diluarea a avut loc în condiții aseptice controlate și validate.
Prospect sursă: AIFA (Agenția italiană pentru medicamente). Conținut publicat în ianuarie 2016. Este posibil ca informațiile prezente să nu fie actualizate.
Pentru a avea acces la cea mai actualizată versiune, este recomandabil să accesați site-ul web AIFA (Agenția italiană pentru medicamente). Declinare de responsabilitate și informații utile.
01.0 DENUMIREA PRODUSULUI MEDICAMENTAL
NIVESTIM 12 MU / 0,2 ML SOLUȚIE PENTRU INJECȚIE / INFUZIE
02.0 COMPOZIȚIA CALITATIVĂ ȘI CANTITATIVĂ
Fiecare ml de soluție injectabilă sau perfuzabilă conține 60 de milioane de unități [UM] (600 mcg) de filgrastim *.
Fiecare seringă preumplută conține 12 milioane de unități (UM) (120 mcg) de filgrastim în 0,2 mL (0,6 mg / mL).
* factor de stimulare a coloniei de granulocite de metionină recombinantă [GCSF]) produs în Escherichia Coli (BL21) cu tehnologie ADN recombinant.
Excipienți cu efect cunoscut
Fiecare ml de soluție conține 50 mg sorbitol.
Pentru lista completă a excipienților, vezi secțiunea 6.1.
03.0 FORMA FARMACEUTICĂ
Soluție injectabilă / perfuzabilă.
Soluție clară, incoloră.
04.0 INFORMAȚII CLINICE
04.1 Indicații terapeutice
Filgrastim este indicat pentru reducerea duratei neutropeniei și a incidenței neutropeniei febrile la pacienții tratați cu chimioterapie citotoxică standard pentru boli maligne (cu excepția leucemiei mieloide cronice și a sindroamelor mielodisplazice) și reducerea duratei neutropeniei la pacienții care suferă de terapie mieloablativă urmată de transplant de măduvă osoasă considerat a fi cu risc crescut de neutropenie severă prelungită.
Siguranța și eficacitatea filgrastimului sunt similare la adulți și copii supuși chimioterapiei citotoxice.
Filgrastim este indicat pentru mobilizarea celulelor progenitoare ale sângelui periferic (PBPC).
La pacienți, copii sau adulți cu neutropenie congenitală, ciclică sau idiopatică severă, cu un număr absolut de neutrofile (ANC) ≤ 0,5 x 109 / L și cu antecedente de infecții severe sau recurente, administrarea pe termen lung a filgrastimului este indicată pentru creșterea neutrofilelor numără și reduce incidența și durata evenimentelor legate de infecție.
Filgrastim este indicat pentru tratamentul neutropeniei persistente (ANC mai mică sau egală cu 1,0 x 109 / l) la pacienții cu infecție HIV avansată pentru a reduce riscul de infecții bacteriene atunci când alte opțiuni de tratament sunt inadecvate.
04.2 Doze și mod de administrare
Terapia cu filgrastim trebuie efectuată numai împreună cu un centru de cancer cu
experiență în tratamentul G-CSF și hematologie și care are echipamentul de diagnostic necesar. Procedurile de mobilizare și afereză trebuie efectuate în colaborare cu un centru de oncologie-hematologie cu experiență acceptabilă în domeniu și unde monitorizarea celulelor progenitoare hematopoietice poate fi efectuată corect.
Dozare
Chimioterapie citotoxică standard
Doza recomandată de filgrastim este de 0,5 MU (5 mcg) / kg / zi. Prima doză de filgrastim trebuie administrată cel puțin 24 de ore după chimioterapia citotoxică.
Doza zilnică de filgrastim ar trebui să continue până când s-a depășit limita de neutrofile așteptată și numărul de nuetrofile a revenit la un nivel normal. După chimioterapia standard pentru tumorile solide, limfoame și leucemii limfoide, durata necesară a tratamentului pentru îndeplinirea acestor criterii ar putea ajunge la 14 zile După tratamentul de inducție și consolidare în leucemia mieloidă acută, durata tratamentului poate fi considerabil mai lungă (până la 38 de zile), în funcție de tipul, doza și modelul chimioterapiei citotoxice utilizate.
La pacienții supuși chimioterapiei citotoxice, o creștere tranzitorie a numărului de neutrofile este de obicei observată la 1-2 zile după inițierea terapiei cu filgrastim. Nadirul așteptat de neutrofile nu a fost depășit și numărul de neutrofile nu a revenit la un nivel normal. Nu este recomandată întreruperea prematură a tratamentului cu filgrastim înainte de atingerea nadirului așteptat de neutrofile.
Pacienții supuși terapiei mieloablative urmate de transplant de măduvă osoasă
Doza inițială recomandată de filgrastim este de 1,0 MU (10 mcg) / kg / zi.
Prima doză de Filgrastim trebuie administrată cel puțin 24 de ore după chimioterapia citotoxică și cel puțin 24 de ore după perfuzia măduvei osoase.
După ce s-a depășit limita de neutrofile, doza zilnică de Filgrastim trebuie titrată pe baza răspunsului la neutrofile după cum urmează:
Mobilizarea PBPC-urilor
Pentru mobilizarea celulelor progenitoare de sânge periferic (PBPC) la pacienții supuși terapiei mielosupresive sau mieloablative urmată de transplant de celule progenitoare de sânge periferic autolog
Doza recomandată de filgrastim pentru mobilizarea PBPC, atunci când este utilizată singură, este de 1,0 MU (10 mcg) / kg / zi timp de 5 până la 7 zile consecutive. Planificarea leucaferezei: 1 sau 2 leucafereze în zilele 5 și 6 sunt adesea suficiente.În alte cazuri, poate fi necesară o leucafereză suplimentară. Administrarea de filgrastim trebuie continuată până la ultima leucafereză.
Doza recomandată de filgrastim pentru mobilizarea PBPC după chimioterapia mielosupresivă este de 0,5 MU (5 mcg) / kg / zi, care trebuie administrată zilnic din prima zi după terminarea chimioterapiei până când s-a depășit limita de neutrofile așteptată și numărul de neutrofile nu a revenit la un nivel normal. Leucafereza trebuie efectuată în perioada în care ANC crește de la 5,0 x 109 / L. La pacienții care nu urmează chimioterapie extinsă, este adesea suficientă o singură leucafereză, iar în alte cazuri se recomandă o leucafereză suplimentară.
Pentru mobilizarea celulelor progenitoare de sânge periferic (PBPC) la donatorii sănătoși înainte de transplantul de celule progenitoare de sânge periferic alogen
Pentru mobilizarea PBPC la donatorii sănătoși, filgrastim trebuie administrat prin injecție subcutanată la o doză de 10 mcg / kg / zi timp de 4 până la 5 zile consecutive. Leucafereza ar trebui să înceapă în ziua 5 și să continue după cum este necesar până în ziua 6 pentru a obține 4 x 106 celule CD34 + / kg greutate corporală a destinatarului.
La pacienții cu neutropenie cronică severă
Neutropenie congenitală: doza recomandată este de 1,2 MU (12 micrograme) / kg / zi ca doză unică sau în doze divizate.
Neutropenie idiopatică sau ciclică: doza inițială recomandată este de 0,5 MU (5 mcg) / kg / zi ca doză unică sau în doze divizate.
Ajustări de dozare: Filgrastim trebuie administrat zilnic până la atingerea numărului de neutrofile și poate fi menținut peste 1,5 x 109 / l. Când se obține răspunsul, trebuie determinată cea mai mică doză eficientă pentru menținerea acestui nivel. Este necesară administrarea zilnică pe termen lung pentru a menține un număr adecvat de neutrofile. După una până la două săptămâni de terapie, doza inițială poate fi dublată sau înjumătățită pe baza răspunsului pacientului. Ulterior, doza poate fi ajustată individual la fiecare 1-2 săptămâni pentru a menține un număr mediu de neutrofile între 1,5 x 109 / L și 10 x 109 / L. La pacienții cu infecții severe, poate fi luat în considerare un program mai rapid de creștere progresivă a dozei. În studiile clinice, 97% dintre respondenți au obținut un răspuns complet la doze ≤ 24 mcg / kg / zi. Siguranța pe termen lung a administrării de filgrastim la doze peste 24 micrograme / kg / zi la pacienții cu neutropenie cronică severă nu a fost demonstrată.
La pacienții cu infecție HIV
Inversarea neutropeniei
Doza inițială recomandată de filgrastim este de 0,1 MU (1 microgram) / kg / zi administrată zilnic cu titrare până la maximum 0,4 MU (4 mcg) / kg / zi până când a fost atinsă și poate avea un număr normal de nuetrofile (ANC> 2,0 x109 / l) să fie întreținute. În studiile clinice,> 90% dintre pacienți au răspuns la aceste doze, realizând inversarea neutropeniei pe o perioadă mediană de 2 zile.
La un număr mic de pacienți (
Menținerea unui număr normal de neutrofile
Când s-a realizat inversarea neutropeniei, trebuie determinată cea mai mică doză eficientă pentru a menține un număr normal de neutrofile. Se recomandă o ajustare inițială a dozei cu o doză alternativă pe zi de 30 MU (300 mcg) / zi. Pot fi necesare ajustări suplimentare ale dozei, în funcție de ANC-ul pacientului, pentru a menține numărul de neutrofile la> 2,0 x 109 / L. În studiile clinice, au fost necesare doze de 30 MU (300 mcg) / zi.1 până la 7 zile pe săptămână pentru a menține ANC> 2,0 x 109 / L, cu o frecvență mediană de administrare de 3 zile pe săptămână. Poate fi necesară administrarea pe termen lung pentru a menține ANC> 2,0 x 109 / L.
Populații speciale
Pacienți vârstnici
Doar un număr mic de pacienți vârstnici au fost incluși în studiile clinice cu filgrastim. Nu au fost efectuate studii specifice la această populație de pacienți. Prin urmare, nu se pot face recomandări specifice de dozare pentru acești pacienți.
Pacienți cu insuficiență renală sau hepatică
Studiile efectuate cu filgrastim la pacienți cu insuficiență renală sau hepatică severă arată că profilul său farmacocinetic și farmacodinamic este similar cu cel observat la subiecții sănătoși. În aceste cazuri, nu este necesară ajustarea dozelor.
Pacienți copii cu neutropenie cronică severă (SCN) și boli maligne
În studiile clinice, șaizeci și cinci la sută dintre pacienții tratați pentru un SCN au avut sub 18 ani. În această grupă de vârstă, incluzând în principal pacienții cu neutropenie congenitală, s-a demonstrat eficacitatea.Nu au fost observate diferențe în profilurile de siguranță ale pacienților pediatrici tratați pentru neutropenie cronică severă.
Datele din studiile clinice efectuate la copii și adolescenți indică faptul că siguranța și eficacitatea filgrastimului sunt similare la adulți și copii supuși chimioterapiei citotoxice.
Recomandările de dozare la copii și adolescenți sunt identice cu recomandările valabile pentru adulții supuși chimioterapiei citotoxice mielosupresive.
Mod de administrare
Chimioterapie citotoxică standard
Filgrastim poate fi administrat prin injecție subcutanată zilnică sau perfuzie intravenoasă zilnică diluată în soluție injectabilă de glucoză 50 mg / ml (5%) timp de 30 de minute (vezi pct. 6.6 din instrucțiunile de diluare). În majoritatea cazurilor, este preferabilă ruta subcutanată. Există dovezi dintr-un studiu cu doză unică că utilizarea intravenoasă poate reduce durata efectului. Nu este clară relevanța clinică a acestei constatări pentru administrarea de doze multiple. Alegerea căii de administrare trebuie să se bazeze pe starea clinică a fiecărui pacient. În studiile clinice randomizate, s-au utilizat doze subcutanate de 230 mcg / m2 / zi (4,0 până la 8,4 mcg / kg / zi).
Pacienții supuși terapiei mieloablative urmate de transplant de măduvă osoasă
Filgrastim se administrează prin perfuzie intravenoasă de 30 de minute sau prin perfuzie intravenoasă sau prin perfuzie subcutanată continuă 24 de ore. Filgrastim trebuie diluat în 20 ml dintr-o soluție perfuzabilă de glucoză 50 mg / ml (5%) (vezi pct. 6.6).
Mobilizarea PBPC-urilor
Pentru mobilizarea celulelor progenitoare de sânge periferic (PBPC) la pacienții supuși terapiei mielosupresive sau mieloablative urmată de transplant de celule progenitoare de sânge periferic autolog, doza recomandată de filgrastim prin perfuzie subcutanată continuă poate fi administrată timp de 24 de ore sau prin injecție subcutanată zilnică unică pentru 5-7 zile consecutive. Pentru perfuzie, filgrastim trebuie diluat în 20 ml dintr-o soluție injectabilă de glucoză 50 mg / ml (5%) (vezi pct. 6.6).
Infecția NCG / HIV
Injecție subcutanată.
Pentru instrucțiuni privind manipularea medicamentului înainte de utilizare, consultați secțiunea 6.6.
04.3 Contraindicații
Hipersensibilitate la substanța activă sau la oricare dintre excipienții menționați la punctul 6.1.
04.4 Avertismente speciale și precauții adecvate pentru utilizare
Avertismente speciale
Filgrastim nu trebuie utilizat pentru a crește doza de chimioterapie citotoxică dincolo de regimul de dozare standard.
Filgrastim nu trebuie utilizat la pacienții cu neutropenie congenitală severă (sindrom Kostman) cu anomalii citogenetice.
Au fost raportate reacții de hipersensibilitate, inclusiv reacții anafilactice, care au apărut la inițierea sau ulterior tratamentului la pacienții tratați cu filgrastim. Întrerupeți definitiv tratamentul cu filgrastim la pacienții cu hipersensibilitate semnificativă clinic.Nu administrați filgrastim la pacienții cu antecedente de hipersensibilitate la filgrastim sau pegfilgrastim.
Ca și în cazul tuturor proteinelor terapeutice, există un risc potențial de imunogenitate. Probabilitatea de a genera anticorpi împotriva filgrastimului este în general scăzută. Dezvoltarea anticorpilor de legare este de așteptat cu toți biologicii;
Proliferarea celulelor maligne
GCSF poate promova proliferarea celulelor mieloide in vitro și se pot observa efecte similare in vitro pe unele celule non-mieloide.
Siguranța și eficacitatea administrării Filgrastim la pacienții cu sindrom mielodisplazic sau leucemie mieloidă cronică nu au fost demonstrate.
Filgrastim nu este indicat în astfel de situații. O atenție deosebită trebuie acordată diagnosticului diferențial între transformarea exploziei în leucemia mieloidă cronică și leucemia mieloidă acută.
Datorită datelor limitate privind siguranța și eficacitatea, filgrastim trebuie administrat cu precauție la pacienții cu LMA secundară.
Siguranța și eficacitatea administrării de filgrastim la pacienții de vârstă de novo și citogenetică favorabilă [t (8; 21), t (15; 17) și inv] nu au fost demonstrate.
Alte precauții speciale
Monitorizarea densității osoase poate fi indicată la pacienții cu osteoporoză de bază care urmează un tratament continuu cu filgrastim mai mult de 6 luni.
Au fost raportate reacții adverse pulmonare rare (> 0,01% și pneumonie interstițială după administrarea de G-CSF. Pacienții cu antecedente recente de infiltrare pulmonară sau pneumonie pot prezenta un risc crescut. Apariția semnelor pulmonare precum tuse, febră și dispnee în asocierea cu semne radiologice de infiltrare pulmonară și deteriorarea funcției pulmonare pot fi semne preliminare ale sindromului de detresă respiratorie la adulți (SDRA). Filgrastim trebuie întrerupt și trebuie inițiat tratamentul adecvat.
S-a raportat sindromul de scurgeri capilare după administrarea factorului stimulator al coloniilor de granulocite și se caracterizează prin hipotensiune arterială, hipoalbuminemie, edem și hemoconcentrare. Pacienții care dezvoltă simptome ale sindromului de scurgeri capilare ar trebui să fie monitorizați îndeaproape și să primească tratament simptomatic standard, care poate include necesitatea terapiei intensive (vezi pct. 4.8).
Precauții speciale la pacienții cu cancer
Leucocitoza
Numărul de celule albe din sânge de 100 x 109 / L sau mai mare a fost observat la mai puțin de 5% dintre pacienții tratați cu filgrastim la doze peste 0,3 MU / kg / zi (3 mcg / kg / zi). Nu au fost observate reacții adverse direct atribuibile acestui grad de leucocitoză. Cu toate acestea, având în vedere riscurile potențiale asociate cu leucocitoza severă, în timpul terapiei cu filgrastim trebuie efectuată o monitorizare regulată a numărului de globule albe din sânge. Tratamentul cu Filgrastim trebuie oprit imediat dacă numărul de celule albe din sânge depășește 50 x 109 / l după nadirul așteptat. Cu toate acestea, în perioada de administrare a filgrastimului pentru mobilizarea PBPC, tratamentul trebuie oprit sau doza trebuie redusă dacă numărul de globule albe depășește 70 x 109 / l.
Riscuri asociate chimioterapiei cu doze mari
Trebuie acordată o atenție deosebită tratamentului pacienților cu chimioterapie cu doze mari, deoarece nu a fost demonstrat un răspuns tumoral mai favorabil și deoarece administrarea chimioterapiei cu doze mari poate crește efectele toxice, inclusiv efecte cardiace, pulmonare, neurologice și dermatologice. (consultați rezumatul caracteristicilor produsului agenților chimioterapeutici utilizați).
Tratamentul cu filgrastim singur nu previne trombocitopenia și anemia după chimioterapia mielosupresivă. Ca urmare a posibilității de a primi doze mai mari de chimioterapie (de exemplu, doze complete conform regimului de dozare prescris), pacientul poate fi expus unui risc crescut de trombocitopenie și anemie. Prin urmare, se recomandă verificarea periodică a numărului de trombocite și a hematocritului. Trebuie acordată o atenție deosebită în timpul administrării, atât singure, cât și în combinație, a agenților chimioterapeutici cunoscuți că induc trombocitopenie severă.
S-a demonstrat că utilizarea PBPC-urilor mobilizate cu filgrastim reduce severitatea și durata trombocitopeniei după chimioterapie mielosupresivă sau mieloablativă.
Splenomegalie
Cazuri de splenomegalie și ruptură splenică au fost raportate neobișnuit după administrarea de filgrastim. Unele cazuri de ruptură splenică au fost fatale. Subiecții care primesc filgrastim și care raportează durerea abdominală superioară stângă și / sau durerea la nivelul umărului trebuie evaluați pentru mărirea splenică sau ruptura splenică.
Alte precauții speciale
Efectul filgrastimului la pacienții cu progenitori mieloizi reduși semnificativ nu a fost studiat. Pentru a crește numărul de neutrofile, filgrastimul acționează în primul rând asupra precursorilor neutrofilelor. infiltrarea tumorii a măduvei osoase), răspunsul neutrofilelor poate fi redus.
Au fost raportate cazuri de boală grefă versus gazdă, GvHD și deces la pacienții tratați cu G-CSF după transplant alogen de măduvă osoasă (vezi pct. 5.1).
Efectul filgrastimului asupra bolii GvHD nu a fost definit.
Creșterea activității hematopoietice a măduvei osoase ca răspuns la terapia cu factor de creștere a fost asociată cu constatări tranzitorii pozitive ale imaginii osoase. Acest lucru ar trebui luat în considerare la interpretarea rapoartelor osoase.
Precauții speciale la pacienții supuși mobilizării celulelor progenitoare a sângelui periferic.
Mobilizare
Nu există studii radiologice prospective care să compare cele două metode de mobilizare recomandate (filgrastim singur sau în combinație cu chimioterapie mielosupresivă) în aceeași populație de pacienți. Gradul de variabilitate între pacienți individuali și între testele de laborator ale celulelor CD34 + demonstrează dificultatea de a compara diferitele este dificil să se recomande o metodă optimă. Alegerea metodei de mobilizare trebuie evaluată în raport cu obiectivele individuale de tratament ale pacientului.
Expunerea anterioară la agenți citotoxici
La pacienții pre-tratați extensiv cu terapie mielosupresivă este posibil ca mobilizarea PBPC să nu fie suficientă pentru a obține numărul minim recomandat de celule (2,0 x 106 celule CD34 + / kg) sau ca accelerația recuperării trombocitelor să fie mai puțin marcată.
Unii agenți citotoxici prezintă o toxicitate deosebită asupra celulelor progenitoare hematopoietice și pot contracara mobilizarea acestora. Substanțe precum melfalan, carmustină (BCNU) și carboplatină, dacă sunt administrate pentru o perioadă prelungită înainte de mobilizarea celulelor progenitoare, pot reduce numărul de celule colectate. Cu toate acestea, administrarea de melphalan, carboplatină sau BCNU în combinație cu filgrastim s-a dovedit a fi eficientă în mobilizarea celulelor progenitoare. Dacă este planificat un transplant de celule progenitoare de sânge periferic, mobilizarea celulelor stem ar trebui planificată în faza inițială a tratamentului intenționat al pacientului. O atenție deosebită trebuie acordată numărului de celule progenitoare mobilizate la astfel de pacienți înainte de administrarea chimioterapiei cu doze mari. Dacă colectarea celulelor este inadecvată în conformitate cu criteriile de evaluare indicate anterior, ar trebui luate în considerare tratamente alternative care nu necesită utilizarea celulelor progenitoare.
Evaluarea colecției de celule progenitoare
În evaluarea cantitativă a celulelor progenitoare obținută la pacienții tratați cu filgrastim, trebuie acordată o atenție deosebită metodei de enumerare. Rezultatele numărului de celule CD34 + prin citometrie în flux variază în funcție de metodologia utilizată; prin urmare, numerele derivate din studii efectuate în alte laboratoare trebuie interpretate cu prudență.
Analiza statistică a relației dintre numărul de celule CD34 + reinfuzate și rata de recuperare a trombocitelor după chimioterapie cu doze mari indică o relație complexă, dar constantă.
Recomandarea de a colecta un număr minim de 2,0 x 106 celule CD34 + / kg se bazează pe experiența publicată, ceea ce indică faptul că recuperarea hematologică este astfel adecvată. Sumele mai mari decât numărul minim indicat par a fi legate de recuperarea mai rapidă, sumele mai mici la recuperarea mai lentă.
Precauții speciale la donatorii sănătoși supuși mobilizării celulelor progenitoare a sângelui periferic
Mobilizarea PBPC nu are niciun beneficiu clinic direct la donatorii sănătoși și trebuie luată în considerare numai în scopul transplantului de celule stem alogene.
Mobilizarea PBPC ar trebui luată în considerare numai la donatorii care îndeplinesc criteriile de eligibilitate clinice și de laborator normale pentru donarea de celule stem, acordând o atenție deosebită parametrilor hematologici și prezenței bolilor infecțioase.
Siguranța și eficacitatea filgrastim nu au fost evaluate la donatorii sănătoși cu vârsta de 60 de ani.
Trombocitopenie tranzitorie (trombocite
Dacă este necesară mai mult de o leucafereză, donatorii cu trombocite
Leucafereza nu trebuie efectuată la donatorii în tratament anticoagulant sau care au cunoscut modificări ale hemostazei.
Administrarea de filgrastim trebuie întreruptă sau doza trebuie redusă dacă numărul de globule albe ajunge la> 70 x109 / l.
Donatorii care primesc G-CSF pentru mobilizarea PBPC ar trebui monitorizați până când parametrii hematologici sunt normalizați.
Modificări citogene tranzitorii au fost observate după utilizarea G-CSF la donatorii sănătoși. Semnificația acestor modificări este necunoscută.
Urmărirea siguranței pe termen lung la donatori este în curs de desfășurare. Cu toate acestea, riscul dezvoltării unei clone celulare mieloide maligne nu poate fi exclus. Se recomandă ca centrul de afereză să efectueze înregistrarea și screeningul sistematic al donatorilor de celule stem pentru a asigura monitorizarea siguranței pe termen lung.
După administrarea G-CSF, splenomegalie în general asimptomatică și, în cazuri foarte rare, ruptura splinei a fost frecvent observată la donatorii sănătoși (și la pacienți). Unele cazuri de rupere a splinei au fost fatale. Prin urmare, volumul splinei trebuie verificat cu atenție (de exemplu, prin examinare fizică, ultrasunete). Diagnosticul de rupere a splinei trebuie luat în considerare la donatori și / sau pacienți cu dureri abdominale superioare stângi sau dureri ale omoplatului.
În experiența de după punerea pe piață, au fost raportate foarte rar evenimente adverse pulmonare (hemoptizie, hemoragie pulmonară, infiltrare pulmonară, dispnee și hipoxie) la donatorii normali după utilizarea altor medicamente care conțin filgrastim. trebuie luat în considerare tratamentul cu filgrastim și trebuie acordată asistența medicală necesară.
Precauții speciale la primitorii de celule progenitoare ale sângelui periferic alogen mobilizat cu filgrastim
Datele actuale indică faptul că interacțiunile imunologice dintre PBPC-urile alogene și beneficiarul pot fi asociate cu un risc crescut de boală acută și cronică de grefă versus gazdă (GvHD) comparativ cu transplantul de măduvă osoasă.
Precauții speciale la pacienții cu neutropenie cronică severă (SCN)
Numărul complet de sânge
Numărul de trombocite trebuie monitorizat frecvent, în special în primele câteva săptămâni de tratament cu filgrastim. La pacienții care dezvoltă trombocitopenie, adică cu trombocite, trebuie avută în vedere întreruperea intermitentă a tratamentului sau reducerea dozei de filgrastim.
Pot apărea alte modificări ale imaginii sanguine, inclusiv anemie și creșteri tranzitorii ale progenitorilor mieloizi, care necesită o monitorizare atentă a numărului de sânge.
Transformarea în leucemie sau sindrom mielodisplazic
O atenție deosebită trebuie acordată diagnosticului diferențial între neutropenia cronică severă și alte boli hematologice, cum ar fi anemia aplastică, mielodispalsia și leucemia mieloidă. Înainte de începerea tratamentului, trebuie efectuată o hemoleucogramă completă, cu număr diferențiat și trombocite, precum și o evaluare a morfologiei măduvei osoase și a unui cariotip.
S-au observat sindroame mielodisplazice (SMD) sau leucemie la un număr mic (aproximativ 3%) de pacienți cu neutropenie cronică severă tratați cu filgrastim în studiile clinice. Acest lucru a fost observat doar la pacienții cu neutropenie congenitală. SMD și leucemia sunt complicații naturale ale bolii și nu trebuie luate în considerare cu certitudine în ceea ce privește tratamentul cu filgrastim. Anomalii, inclusiv monozomia 7, au fost constatate ulterior la aproximativ 12% dintre pacienții cu citogenetică normală la momentul inițial în timpul testelor repetate de rutină. Dacă pacienții cu neutropenie cronică severă dezvoltă anomalii citogenetice, trebuie luate în considerare cu atenție riscurile și beneficiile continuării tratamentului cu filgrastim; Administrarea de filgrastim trebuie întreruptă în cazul în care apare SMD sau leucemie. În prezent nu se știe dacă tratamentul pe termen lung al pacienților cu neutropenie cronică severă poate predispune pacienții la anomalii citogenetice, SMD sau transformare leucemică. La acești pacienți, se recomandă analize morfologice și citogenetice ale măduvei osoase la intervale regulate (aproximativ la fiecare 12 luni).
Splenomegalie
Cazuri de splenomegalie și ruptură splenică au fost raportate neobișnuit după administrarea de filgrastim. Unele cazuri de ruptură splenică au fost fatale. Subiecții care primesc filgrastim și care raportează durerea abdominală superioară stângă și / sau durerea la nivelul umărului trebuie evaluați pentru mărirea splenică sau ruptura splenică.
Alte precauții speciale
Cauzele neutropeniei tranzitorii, cum ar fi infecțiile virale, trebuie excluse.
Splenomegalia este un efect direct al tratamentului cu filgrastim. Splenomegalie palpabilă a fost observată la 31% dintre pacienți în studiile clinice. Creșteri de volum, măsurate radiologic, au fost observate devreme în timpul terapiei cu filgrastim și au arătat o tendință de stabilizare. S-a observat că reducerea dozelor încetinește sau oprește progresia splenomegaliei și a fost necesară o splenectomie la 3% dintre pacienți. Volumul splinei trebuie verificat în mod regulat. Palparea abdominală este suficientă pentru a detecta creșteri anormale ale volumului.
Hematuria / proteinuria a apărut la un număr mic de pacienți. Analiza urinei trebuie făcută la intervale regulate pentru a detecta astfel de evenimente.
Siguranța și eficacitatea la nou-născuți și la pacienții cu neutropenie autoimună nu au fost demonstrate.
Precauții speciale la pacienții infectați cu HIV
Numărul complet de sânge
Numărul absolut de neutrofile (ANC) trebuie monitorizat frecvent, în special în primele câteva săptămâni de tratament cu filgrastim. Unii pacienți pot răspunde foarte rapid și cu o creștere semnificativă a numărului de neutrofile la doza inițială de filgrastim. Se recomandă ca ANC să fie determinat zilnic în primele 2-3 zile de administrare a filgrastimului. Ulterior, se recomandă ca ANC să fie determinat cel puțin de două ori pe săptămână în primele 2 săptămâni și o dată pe săptămână sau la două săptămâni după aceea. în timpul terapiei de întreținere. Cu administrarea intermitentă de 30 MU (300 mcg) / zi de filgrastim, pot apărea fluctuații mari în timp ale ANC. Pentru a determina valoarea minimă sau mai mică a ANC a unui pacient, se recomandă prelevarea de probe de sânge destinate determinării ANC. imediat înainte de administrarea intenționată de filgrastim.
Riscuri asociate cu doze mari de medicamente mielosupresive
Tratamentul cu filgrastim singur nu previne trombocitopenia și anemia după terapia mielosupresivă. Deoarece pot fi administrate doze mai mari sau mai multe dintre aceste medicamente odată cu utilizarea filgrastimului, pacientul poate prezenta un risc crescut de trombocitopenie sau anemie. Se recomandă monitorizarea regulată a hematocritului (vezi mai sus).
Infecții și tumori maligne care cauzează mielosupresie
O neutropenie se poate datora infiltrării măduvei osoase din infecții oportuniste, cum ar fi Mycobacterium avium complexe sau la neoplasme maligne, cum ar fi limfoamele. La pacienții cu infecții cunoscute de infiltrare a măduvei osoase sau tumori maligne, trebuie luată în considerare tratamentul adecvat al bolii de bază, pe lângă administrarea de filgrastim pentru tratamentul neutropeniei. Efectele filgrastimului asupra neutropeniei datorate infecțiilor cu infiltrare a măduvei osoase sau a tumorilor maligne nu au fost demonstrate în mod concludent.
Splenomegalie
Cazuri de splenomegalie și ruptură splenică au fost raportate neobișnuit după administrarea de filgrastim. Unele cazuri de ruptură splenică au fost fatale. Subiecții care primesc filgrastim și care raportează durerea abdominală superioară stângă și / sau durerea la nivelul umărului trebuie evaluați pentru mărirea splenică sau ruptura splenică.
Precauții speciale în anemia falciformă sau falciformă
Crizele falciforme, în unele cazuri letale, au fost raportate la pacienții cu seceră sau cu celule falciforme tratați cu filgrastim. La pacienții cu trăsături cu celule falciforme sau cu anemie falciformă, medicii trebuie să fie precauți atunci când evaluează utilizarea filgrastimului, care trebuie utilizat numai după o analiză atentă a beneficiilor și riscurilor potențiale.
Excipienți
Nivestim conține sorbitol. Pacienții cu intoleranță ereditară la fructoză nu trebuie să utilizeze acest medicament. De asemenea, conține mai puțin de 1 mmol sodiu (23 mg) pe doză, adică în esență „fără sodiu”.
04.5 Interacțiuni cu alte medicamente și alte forme de interacțiune
Siguranța și eficacitatea filgrastimului administrat în aceeași zi cu chimioterapia citotoxică mielosupresivă nu au fost demonstrate în mod concludent. Deoarece celulele mieloide cu divizare rapidă sunt sensibile la chimioterapia citotoxică mielosupresivă, utilizarea filgrastimului nu este recomandată în această perioadă. ore după chimioterapie. Datele preliminare obținute la un număr mic de pacienți tratați în comun cu filgrastim și 5-fluorouracil indică faptul că neutropenia se poate agrava.
Interacțiunile posibile cu alți factori de creștere hematopoietică și citokine nu au fost încă studiate în studiile clinice.
Deoarece litiul favorizează eliberarea neutrofilelor, este probabil să potențeze efectul filgrastimului. Deși această interacțiune nu a fost studiată în mod formal, nu există dovezi că este dăunătoare.
04.6 Sarcina și alăptarea
Sarcina
Nu există sau există date limitate despre utilizarea filgrastim la femeile gravide.
Studiile la animale au arătat toxicitate asupra funcției de reproducere. La „iepuri a fost observată o„ incidență crescută a avorturilor spontane după expunerea ”la multipli mari de doze clinice
și în prezența toxicității materne (vezi pct. 5.3). Au fost descrise cazuri în literatura de specialitate în care răspândirea placentară a filgrastimului a fost demonstrată la femeile gravide. Filgrastim nu este recomandat în timpul sarcinii.
Timp de hrănire
Nu se știe dacă filgrastimul este excretat în laptele matern uman; prin urmare, filgrastim nu este recomandat la femeile care alăptează. Trebuie luată o decizie dacă întreruperea alăptării sau întreruperea / abținerea tratamentului cu filgrastim, luând în considerare beneficiul alăptării pentru copil și beneficiul terapiei pentru femeie.
Fertilitate
Filgrastim nu a avut niciun efect asupra reproducerii sau fertilității la șobolanii masculi sau femele (vezi pct. 5.3).
04.7 Efecte asupra capacității de a conduce vehicule și de a folosi utilaje
Filgrastim are efecte modeste asupra capacității de a conduce vehicule și de a folosi utilaje.
04.8 Efecte nedorite
Rezumatul profilului de siguranță
În studiile clinice, 183 de pacienți cu cancer și 96 de voluntari sănătoși au fost expuși la Nivestim.
Profilul de siguranță al filgrastimului observat în aceste studii clinice a fost în concordanță cu cel raportat pentru produsul de referință utilizat în aceste studii.
În studiile clinice la pacienții cu cancer, reacția adversă atribuită filgrastimului la cea mai frecventă doză recomandată a fost durerea musculo-scheletică, ușoară sau moderată la 10% și severă la 3% dintre pacienți.
De asemenea, a fost raportată boala de reacție a grefei versus gazdă (GvHD) (vezi mai jos).
În mobilizarea celulelor stem circulante periferice (PBPC) la donatorii sănătoși, cea mai frecventă reacție adversă raportată a fost durerea musculo-scheletală. Leucocitoza a fost observată la donatori și trombocitopenia după filgrastim și leucafereza a fost observată și la donatori. De asemenea, au fost raportate splenomegalie și ruptură splenică. Unele cazuri de ruptură splenică au fost fatale.
La pacienții cu neutropenie cronică severă (SCN), cele mai frecvente reacții adverse atribuite filgrastimului au fost durerea osoasă, durerea musculo-scheletală generală și splenomegalia.
Sindromul de scurgeri capilare, care poate pune viața în pericol dacă tratamentul este întârziat, a fost raportat mai puțin frecvent (≥1 / 1000 până la
În studiile clinice la pacienții cu HIV, singurele reacții adverse care au fost considerate legate în mod unic de administrarea de filgrastim au fost durerea musculo-scheletică, durerea osoasă ușoară până la moderată și mialgia. Incidența acestor reacții a fost similară cu cea raportată la pacienții cu cancer.
Tabelul reacțiilor adverse
Reacțiile adverse enumerate mai jos și frecvența acestora au fost observate după tratamentul cu filgrastim conform datelor publicate.
Frecvențele reacțiilor adverse sunt definite în conformitate cu următoarele convenții:
Foarte frecvente: ≥1 / 10
Frecvente: ≥1 / 100 ani
Mai puțin frecvente: ≥1 / 1.000 ani
Rare: ≥1 / 10.000 y
Foarte rar:
Necunoscut: nu poate fi estimat din datele disponibile
În cadrul fiecărei grupe de frecvență, reacțiile adverse sunt prezentate în ordinea descrescătoare a severității.
La bolnavii de cancer
Donatorii sănătoși supuși mobilizării celulelor progenitoare din sângele periferic
La pacienții cu neutropenie cronică severă (SCN)
La pacienții cu HIV
Descrierea reacțiilor adverse selectate
GvHD și decese au fost raportate la pacienții cărora li s-a administrat G-CSF după transplant alogen de măduvă osoasă (vezi pct. 5.1).
Au fost raportate cazuri de sindrom de scurgere capilară după punerea pe piață, cu utilizarea factorilor de stimulare a coloniilor de granulocite, care au apărut în general la pacienții cu boală malignă avansată, sepsis, care luau mai multe medicamente chimioterapice sau erau supuși aferezei (vezi pct. 4.4).
La bolnavii de cancer
Durerea musculo-scheletică este de obicei controlată cu analgezice standard. Reacțiile adverse mai puțin frecvente includ anomalii urinare cu disurie predominant ușoară sau moderată.
În studiile randomizate, controlate cu placebo, filgrastim nu a crescut incidența evenimentelor adverse asociate cu chimioterapia citotoxică. , cefalee, erupție pe tuse, durere în piept, slăbiciune generală, durere în gât, constipație și durere nespecificată.
S-au raportat creșteri reversibile, dependente de doză și, de obicei, ușoare până la moderate ale lactatului dehidrogenazei, fosfatazei alcaline, acidului uric la 50%, 35%, 25% și 10% dintre pacienții tratați cu filgrastim la dozele recomandate, respectiv ser și gamma -glutamiltranspeptidaza.
Ocazional au fost raportate creșteri tranzitorii ale tensiunii arteriale, care nu necesită tratament clinic.
Tulburări vasculare, inclusiv boala veno-ocluzivă și modificări ale volumului fluidelor, au fost raportate ocazional la pacienții tratați cu doze mari de chimioterapie, urmate de transplant autolog de măduvă osoasă. Nu a fost demonstrată o relație de cauzalitate cu filgrastim.
Au fost raportate evenimente adverse rare de vasculită cutanată la pacienții tratați cu filgrastim. Nu se cunoaște mecanismul vasculitei la pacienții tratați cu filgrastim.
Au fost descrise cazuri ocazionale de sindrom Sweet (dermatoză febrilă acută). Cu toate acestea, întrucât un procent semnificativ dintre acești pacienți au fost diagnosticați cu leucemie, o afecțiune cunoscută a fi asociată cu sindromul Sweet, nu a fost demonstrată o relație cauzală cu filgrastim.
În cazuri individuale a fost raportată o „exacerbare a” poliartritei reumatoide.
Au fost raportate cazuri rare de evenimente adverse pulmonare, inclusiv pneumonie interstițială, edem pulmonar și infiltrații pulmonare cu insuficiență respiratorie sau sindrom de detresă respiratorie la adulți (ARDS), care pot fi letale (vezi pct. 4.4).
Reacții alergice: au fost raportate reacții de tip alergic, inclusiv anafilaxie, erupții cutanate, urticarie, angioedem, dispnee și hipotensiune arterială, care au apărut la pacienții care au primit primul sau ulterior tratament cu filgrastim. În general, rapoartele au fost mai frecvente după administrare. În unele cazuri, simptomele au recurent la reluarea tratamentului, sugerând o relație cauzală La pacienții care prezintă o reacție alergică severă la filgrastim, tratamentul trebuie întrerupt definitiv.
Au fost raportate cazuri izolate de crize de celule falciforme la pacienții cu anemie falciformă sau falciformă (vezi pct. 4.4). Din datele clinice, frecvența este estimată ca fiind neobișnuită.
Pseudogout a fost raportat la pacienții cu cancer tratați cu filgrastim.
Donatorii sănătoși supuși mobilizării celulelor progenitoare din sângele periferic
Leucocitoza (număr de globule albe (globule albe)> 50 x 109 / l) a fost raportată la 41% dintre donatori și trombocitopenie (trombocite
Au fost raportate creșteri tranzitorii și ușoare ale fosfatazei alcaline, LDH, SGOT și acid uric la donatorii sănătoși tratați cu filgrastim care nu au avut sechele clinice.
O „exacerbare a simptomelor artritice a fost raportată foarte rar.
Reacții alergice severe au fost raportate foarte rar.
Cefaleea, considerată a fi declanșată de filgrastim, a fost raportată în studii efectuate cu donatori sănătoși de PBPC.
Cazuri asimptomatice de splenomegalie și cazuri foarte rare de ruptură splenică au fost raportate frecvent la donatorii sănătoși și la pacienții după administrarea factorilor de stimulare a coloniei de granulocite (G-CSF) (vezi pct. 4.4).
Evenimentele adverse pulmonare la donatorii sănătoși (hemoptizie, hemoragie pulmonară, infiltrate pulmonare, dispnee și hipoxie) au fost raportate foarte rar în experiența după punerea pe piață a altor medicamente filgrastim (vezi pct. 4.4).
La pacienții cu neutropenie cronică severă (SCN)
Au fost raportate reacții adverse legate de terapia cu filgrastim la pacienții cu SCN și pentru unii dintre ei frecvența lor tinde să scadă în timp.
Reacțiile adverse includ splenomegalie care poate fi progresivă într-o minoritate de cazuri și trombocitopenie. Cefaleea și diareea au fost raportate de obicei la mai puțin de 10% dintre pacienți la scurt timp după inițierea terapiei cu filgrastim. Au fost raportate și anemie și epistaxis.
Au fost raportate creșteri tranzitorii ale serului fără simptome clinice pentru acidul uric, lactat dehidrogenază și fosfatază alcalină. Au fost raportate, de asemenea, scăderi tranzitorii și moderate ale glicemiei fără post.
Printre reacțiile adverse legate probabil de terapia cu filgrastim care au apărut în mod normal în artralgie, alopecie, osteoporoză și erupții cutanate.
Vasculita cutanată a fost raportată la 2% dintre pacienții cu SCN după utilizare prelungită. Au existat doar câteva cazuri de proteinurie / hematurie.
La pacienții cu HIV
S-a raportat că splenomegalia este secundară terapiei cu filgrastim în hipersplenism și niciun pacient nu a suferit splenectomie. Relația cu tratamentul cu filgrastim este necunoscută, deoarece splenomegalia este frecventă la pacienții infectați cu HIV și este prezentă în grade diferite la majoritatea pacienților cu SIDA.
Populația pediatrică
Datele din studiile clinice efectuate la copii și adolescenți indică faptul că siguranța și eficacitatea filgrastimului sunt similare atât la adulți, cât și la copiii cărora li se administrează chimioterapie citotoxică, sugerând că nu există diferențe legate de vârstă în farmacocinetica filgrastimului. Singura reacție adversă raportată în mod constant a fost durerea musculo-scheletică, care nu diferă de experiența din populația adultă.
Nu există date suficiente pentru a evalua în continuare utilizarea filgrastim la copii și adolescenți.
Alte populații speciale
Utilizare geriatrică
În general, nu s-au observat diferențe de siguranță sau eficacitate între subiecții cu vârsta peste 65 de ani și adulții mai tineri (> 18 ani) cărora li s-a administrat chimioterapie citotoxică, iar experiența clinică nu a identificat diferențe de răspuns între pacienții adulți mai în vârstă și mai tineri. Nu există date suficiente pentru a evalua utilizarea filgrastimului la persoanele geriatrice pentru celelalte indicații aprobate de filgrastim.
Pacienți copii și adolescenți cu neutropenie cronică severă (SNG)
Au fost raportate cazuri de scădere a densității osoase și de osteoporoză la pacienții copii și adolescenți cu neutropenie cronică severă care au primit tratament cronic cu filgrastim. Frecvența este estimată ca fiind „frecventă” din datele din studiile clinice.
Raportarea reacțiilor adverse suspectate
Raportarea reacțiilor adverse suspectate care apar după autorizarea medicamentului este importantă, deoarece permite monitorizarea continuă a raportului beneficiu / risc al medicamentului. Profesioniștii din domeniul sănătății sunt rugați să raporteze orice reacții adverse suspectate prin intermediul sistemului național de raportare. "Anexa V .
04.9 Supradozaj
Efectele supradozajului cu filgrastim nu au fost demonstrate.
Întreruperea tratamentului cu filgrastim are ca rezultat de obicei o scădere cu 50% a neutrofilelor circulante în decurs de 1-2 zile și revenirea la normal în decurs de 1-7 zile.
05.0 PROPRIETĂȚI FARMACOLOGICE
05.1 Proprietăți farmacodinamice
Grupa farmacoterapeutică: imunostimulant, factori de stimulare a coloniilor.
Codul ATC: L03AA02.
Nivestim este un medicament biosimilar. Informații detaliate sunt disponibile pe site-ul Agenției Europene pentru Medicamente www.ema.europa.eu
G-CSF uman este o glicoproteină care reglează producția și eliberarea neutrofilelor funcționale din măduva osoasă. Nivestim, care conține r-metHuG-CSF (filgrastim), induce o creștere semnificativă a numărului de neutrofile din sângele periferic și o creștere mai puțin marcată a monocitelor în decurs de 24 de ore. La unii pacienți cu neutropenie cronică severă, filgrastimul poate induce chiar o ușoară creștere a numărului de eozinofile și bazofile circulante de la momentul inițial; unii dintre acești pacienți pot prezenta eozinofilie sau bazofilie chiar înainte de tratament. La dozele recomandate, creșterea numărului de neutrofile este dependentă de doză.După cum s-a demonstrat în analizele efectuate, neutrofilele produse ca răspuns la filgrastim prezintă proprietăți chimiotactice și fagocitare normale sau crescute. La finalizarea tratamentului cu filgrastim, numărul neutrofilelor circulante scade cu aproximativ 50% în decurs de 1-2 zile și atinge nivelurile normale în decurs de 1-7 zile.
Utilizarea filgrastimului la pacienții supuși chimioterapiei citotoxice reduce semnificativ incidența, severitatea și durata nuetropeniei și a neutropeniei febrile. Tratamentul cu filgrastim reduce semnificativ durata neutropeniei febrile, utilizarea antibioticelor și spitalizarea după chimioterapie de inducție în leucemie mieloidă acută sau terapie mieloablativă urmată de transplant de măduvă osoasă. În ambele cazuri, incidența febrei și a infecțiilor documentate nu a fost redusă, durata febrei nu a fost redusă la pacienții supuși terapiei mieloablative urmată de transplantul de măduvă osoasă.
Utilizarea filgrastimului, fie singură, fie după chimioterapie, mobilizează celulele progenitoare hematopoietice în sângele periferic. Aceste celule progenitoare de sânge periferic autolog (PBPC) pot fi colectate și reinfuzate după chimioterapie citotoxică cu doze mari, ca alternativă sau în plus la transplantul de măduva osoasă.Infuzia cu PBPC accelerează recuperarea hematopoietică și astfel reduce durata riscului de complicații sângerante și necesitatea transfuziilor de trombocite.
Beneficiarii celulelor progenitoare ale sângelui alogen periferic mobilizate cu filgrastim, în comparație cu primitorii alogeni ai transplantului de măduvă osoasă, au raportat o recuperare hematologică rapidă semnificativă, cu o recuperare semnificativă a timpului fără aport de trombocite.
Un studiu european retrospectiv, în care a fost analizată utilizarea G-CSF după transplantul de măduvă osoasă alogenă la pacienții cu leucemie acută, a indicat un risc crescut de GvHD și mortalitate după administrarea G-CSF (TRM Într-un alt studiu retrospectiv internațional, efectuat la pacienții cu leucemii mieloide acute și cronice, nu s-a observat niciun efect asupra riscului de GvHD, TRM și mortalitate. Într-o meta-analiză a studiilor de transplant alogen, inclusiv rezultatele a nouă studii prospective randomizate, 8 studii retrospective și 1 caz-control studiu, nu s-au observat efecte asupra riscului de GvHD acut, GvHD cronic sau mortalitate precoce legată de tratament.
a Analiza include studii privind transplantul de măduvă osoasă în perioada în cauză; GM-CSF a fost utilizat în unele studii
b Analiza include pacienți cărora li s-a efectuat transplant de măduvă osoasă în perioada în cauză
Utilizarea filgrastimului pentru mobilizarea PBPC la donatorii sănătoși înainte de transplantul alogen PBPC are ca rezultat recuperarea a 4 x 106 celule CD34 + / per greutate corporală de primitor la majoritatea donatorilor după două leucafereze. Donatorilor normali li se administrează o doză de 10 mcg / kg / zi, subcutanat timp de 4-5 zile consecutive.
Utilizarea filgrastimului la pacienți, adulți și copii, cu neutropenie cronică severă (neutropenie congenitală, ciclică și idiopatică severă) induce o creștere semnificativă a numărului absolut de neutrofile din sângele periferic și o reducere a episoadelor infecțioase și a evenimentelor conexe.
Utilizarea filgrastimului la pacienții infectați cu HIV menține numărul de neutrofile la niveluri normale și astfel permite administrarea medicamentelor antivirale și / sau mielosupresive conform intenției. Nu există dovezi că pacienții infectați cu HIV și tratați cu filgrastim prezintă replicare crescută a HIV.
Așa cum s-a observat cu alți factori de creștere hematopoietici, G-CSF arată, de asemenea in vitro un efect stimulator asupra celulelor endoteliale umane.
Eficacitatea și siguranța Nivestim au fost studiate în studii randomizate, controlate de fază III în cancerul de sân. Nu s-au găsit diferențe relevante între Nivestim și produsul de referință în ceea ce privește durata neutropeniei severe și incidența neutropeniei febrile.
05.2 Proprietăți farmacocinetice
Un studiu randomizat, necriptat, cu doză unică, controlat prin comparator, în duplicat crossover efectuate pe 46 de voluntari sănătoși au arătat că profilul farmacocinetic al Nivestim a fost comparabil cu cel al produsului de referință după tratamentul subcutanat și intravenos.
Într-un alt studiu randomizat, dublu-orb, cu doze multiple, controlat prin comparator, dublu-încrucișat la 50 de voluntari sănătoși, a arătat că profilul farmacocinetic al Nivestim a fost comparabil cu cel al produsului de referință după tratamentul subcutanat.
S-a demonstrat că clearance-ul filgrastim urmează farmacocinetica de ordinul întâi după tratamentul subcutanat și intravenos. Timpul de înjumătățire plasmatică prin eliminare al filgrastimului este de aproximativ 3,5 ore, cu o rată de eliminare de aproximativ 0,6 ml / min / kg. orice acumulare de medicament cu un timp de înjumătățire comparabil. Prin urmare, există o corelație liniară pozitivă între doză și concentrația serică de filgrastim, indiferent dacă este administrat intravenos sau subcutanat. După administrarea subcutanată a dozelor recomandate, concentrațiile serice au fost menținute peste 10 ng / ml timp de 8 - 16 ore. Volumul de distribuție în sânge este de aproximativ 150 ml / kg.
05.3 Date preclinice de siguranță
Filgrastim a fost studiat în studii de toxicitate după doze repetate cu durata de până la 1 an, care au relevat modificări atribuite efectelor farmacologice așteptate, inclusiv leucocite crescute, hiperplazie de măduvă osoasă mieloidă, granulocitopoieză extramedulară și mărire splenică.
Aceste modificări sunt reversibile după întreruperea tratamentului.
Efectele filgrastimului asupra dezvoltării prenatale au fost studiate la șobolani și iepuri. Administrarea intravenoasă (80 micrograme / kg / zi) de filgrastim la iepuri în perioada organogenezei a arătat toxicitate maternă și o creștere a avorturilor spontane, pierderea post-implantare și scăderea dimensiunii medii a așternutului viu și a greutății fetale.
Pe baza datelor raportate pentru un alt produs filgrastim, s-au observat rezultate similare dincolo de „malformațiile fetale crescute la o doză de 100 mcg / kg / zi, o doză de toxicitate maternă corespunzătoare unei„ expuneri sistemice de aproximativ 50-90. pacienți tratați cu doza clinică de 5 mcg / kg / zi. Nivelul la care nu s-a observat niciun efect advers pentru toxicitatea embrion-fetală în acest studiu a fost de 10 mcg / kg / zi, ceea ce a corespuns unei expuneri sistemice de aproximativ 3-5 ori expunerea observată la pacienții tratați cu doza clinică.
La șobolanii gravide, nu s-a observat toxicitate maternă sau fetală la doze peste 575 mcg / kg / zi. Administrarea de filgrastim la descendenții șobolanilor în perioadele peri-natale și de lactație a arătat o întârziere a diferențierii externe și a întârzierii creșterii (≥ 20 mcg / kg / zi) și o rată de supraviețuire ușor redusă (100 mcg / kg / zi). Zi) .
Nu au fost observate efecte asupra fertilității la șobolanii masculi sau femele pentru filgrastim.
06.0 INFORMAȚII FARMACEUTICE
06.1 Excipienți
Acid acetic, glacial
Hidroxid de sodiu
Sorbitol (E420)
Polisorbat 80
Apă pentru preparate injectabile
06.2 Incompatibilitate
Nivestim nu trebuie diluat cu soluții de clorură de sodiu.
Filgrastimul diluat poate fi absorbit de sticlă și materiale plastice, cu excepția cazului în care este diluat într-o soluție perfuzabilă de glucoză de 50 mg / ml (5%) (vezi pct. 6.6).
Acest medicament nu trebuie amestecat cu alte medicamente, cu excepția celor enumerate la punctul 6.6.
06.3 Perioada de valabilitate
Seringă preumplută
30 de luni.
După diluare
Stabilitatea chimică și fizică la utilizare a soluției diluate pentru perfuzie a fost demonstrată timp de 24 de ore la 2 ° C până la 8 ° C. Din punct de vedere microbiologic, produsul trebuie utilizat imediat. Dacă medicamentul nu este utilizat imediat, utilizatorul este responsabil pentru durata și condițiile de păstrare înainte de utilizare; medicamentul poate fi păstrat până la 24 de ore la o temperatură între 2 ° C și 8 ° C, cu excepția cazului în care diluarea are loc în condiții aseptice controlate și validate.
06.4 Precauții speciale pentru depozitare
A se păstra și transporta la frigider (2 ° C - 8 ° C).
Nu înghețați.
Păstrați seringile preumplute în cutie pentru a le proteja de lumină.
Expunerea accidentală la temperaturi de îngheț de până la 24 de ore nu are niciun efect negativ asupra stabilității Nivestim. Seringa preumplută congelată poate fi decongelată și returnată la frigider pentru utilizare ulterioară. Dacă expunerea la temperaturi scăzute este mai mare de 24 de ore sau dacă este înghețată de mai multe ori, Nivestim NU mai trebuie utilizat.
În perioada de valabilitate și pentru uz ambulatoriu, pacientul poate scoate produsul din frigider și îl poate păstra la temperatura camerei (nu peste 25 ° C) pentru o singură dată și până la 7 zile. frigiderul și trebuie aruncat.
Pentru condițiile de depozitare a medicamentului după diluare, vezi pct. 6.3.
06.5 Natura ambalajului imediat și conținutul ambalajului
Seringă preumplută (sticlă tip I) cu ac de injecție (oțel inoxidabil) cu protecție de siguranță a acului, conținând 0,2 ml soluție injectabilă / perfuzabilă.
Dimensiunea ambalajului de 1, 5 sau 10 seringi preumplute.
Este posibil ca nu toate mărimile de ambalaj să fie comercializate.
06.6 Instrucțiuni de utilizare și manipulare
Dacă este necesar, Nivestim poate fi diluat în soluție injectabilă de glucoză 50 mg / ml (5%).
Nu se recomandă niciodată diluarea la o concentrație finală sub 0,2 MU (2 mcg) per ml.
Soluția trebuie inspectată vizual înainte de utilizare. Trebuie utilizate numai soluții clare, fără particule.
La pacienții tratați cu filgrastim diluat la concentrații sub 1,5 MU (15 mcg) per ml, albumina serică umană (HSA) trebuie adăugată la o concentrație finală de 2 mg / ml.
Exemplu: Într-un volum final de injectat de 20 ml, trebuie administrate doze totale de filgrastim sub 30 MU (300 mcg) prin adăugarea a 0,2 ml soluție de albumină umană 20%.
Filgrastim diluat în soluție de glucoză la 50 mg / ml (5%) este compatibil cu sticla și multe materiale plastice, inclusiv PVC, poliolefină (un copolimer de polipropilenă și polietilenă) și polipropilenă.
Nivestim nu conține conservanți. Datorită riscului de contaminare bacteriană, seringile Nivestim sunt de unică folosință. Produsul medicinal neutilizat și deșeurile derivate din acest medicament trebuie eliminate în conformitate cu reglementările locale.
07.0 DEȚINĂTORUL AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
Hospira UK Limited
Queensway
Royal Leamington Spa
Warwickshire CV31 3RW
Regatul Unit
08.0 NUMĂRUL AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
EU / 1/10/631/001
040158014
EU / 1/10/631/002
040158026
EU / 1/10/631/003
040158038
09.0 DATA PRIMEI AUTORIZAȚII SAU REÎNNOIREA AUTORIZAȚIEI
Data primei autorizații: 08 iunie 2010
10.0 DATA REVIZUIRII TEXTULUI
D.CCE mai 2015