Generalitate
Termenul retinita pigmentara (RP) identifică un grup de boli genetice caracterizate prin degenerare progresivă a retinei.

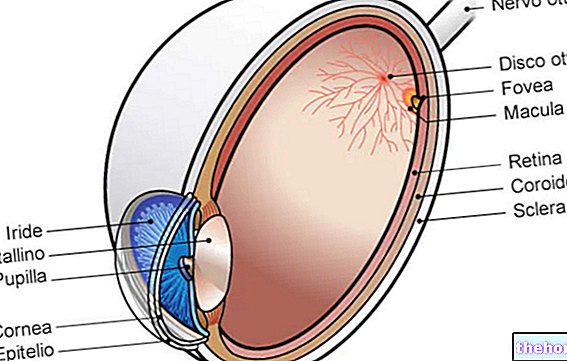
Retinita pigmentară este o distrofie retiniană caracterizată prin pierderea treptată a fotoreceptorilor și disfuncția epiteliului pigmentar, ceea ce înseamnă că retina își reduce progresiv capacitatea de a transmite informații vizuale către creier prin intermediul nervului optic.
Procesul patologic începe cu modificări ale epiteliului pigmentar retinian. Pe măsură ce progresează retinita pigmentară, există o subțire a vaselor de sânge care furnizează retina, care suferă atrofie. La examinarea fundului, depunerile caracteristice sunt detectabile vizual. Pigment retinian ( de aici și numele din boală). Modificările atrofice și leziunile pot implica, de asemenea, nervul optic și, treptat, celulele fotosensibile ale retinei mor.
Pacienții afectați de retinită pigmentară întâmpină inițial probleme de vedere, în special în medii slab iluminate și se plâng de o constricție a câmpului vizual periferic. Viziunea centrală este menținută până la stadiile ulterioare ale bolii, iar rezultatul final poate varia dramatic: multe persoane cu retinită pigmentară își păstrează vederea limitată pe tot parcursul vieții, în timp ce altele își pierd cu totul vederea.
Retinita pigmentară este o boală moștenită, cauzată în principal de modificări genetice transmise de la unul sau de la ambii părinți. Tipul defectului genetic determină ce celule retiniene sunt cele mai implicate în tulburare și face posibilă distincția, din punct de vedere clinic, a diferitelor condiții. Până în prezent, au fost identificate peste 50 de defecte genetice diferite implicate în retinita pigmentară. Anomaliile pot fi transmise de la părinți la descendenți printr-unul din cele trei tipare de moștenire: autosomal recesiv, autosomal dominant sau heterosomal recesiv (X-linked sau X-linked).
Simptome
Pentru informații suplimentare: simptome ale retinitei pigmentare
Retinita pigmentară se găsește de obicei la adolescenți și adulți tineri. Simptomele apar adesea între 10 și 30 de ani, dar diagnosticul poate fi pus în copilăria timpurie sau mult mai târziu în viață.
Simptomele timpurii ale retinitei pigmentare pot include:
- Dificultăți de a vedea noaptea (orbire nocturnă) sau în condiții de lumină slabă
- Adaptare lentă de la viziunea în întuneric la cea din lumină și invers;
- Îngustarea câmpului vizual și pierderea vederii periferice;
- Sensibilitate la lumină și strălucire.
Unele simptome depind de tipul de fotoreceptori implicați. Lansetele sunt responsabile pentru viziunea alb-negru, în timp ce conurile vă permit să deosebiți culorile.
În majoritatea cazurilor de retinită pigmentară, sunt implicate mai întâi tijele. Cu toate acestea, în formele care evoluează rapid, conurile pot fi, de asemenea, afectate într-un stadiu incipient.
Lansetele sunt concentrate în părțile exterioare ale retinei și sunt activate de lumina slabă, astfel încât degenerarea lor afectează viziunea periferică și cea nocturnă. Dacă sunt implicate conuri, este posibil să experimentați pierderea percepției culorii și a vederii centrale.
Predominanța fotoreceptorilor implicați este determinată de defectul particular prezent în structura genetică a pacientului.
Adesea, primul simptom al retinitei pigmentare este orbirea nocturnă (sau noctopia). Unii oameni consideră că au nevoie de tot mai mult timp pentru a se adapta la diferențele de lumină pe măsură ce trec de la o zonă bine luminată la una mai întunecată. O formă tipică de pierdere a vederii induce îngustarea vederii periferice (viziunea prin tunel sau telescop); acest model se numește scotom inelar. Uneori, acest fenomen poate lipsi în stadiile incipiente, dar se observă când individul strică adesea peste obiecte sau este implicat într-un accident de circulație. Când pierderea vederii implică pacienții din zona centrală a retinei (numită și distrofie maculară) întâmpină dificultăți la citire și la lucrări detaliate care necesită concentrare pe un singur obiect, cum ar fi infilarea unui fir prin ochiul unui ac Mulți pacienți raportează că au văzut sclipiri de lumină (fotopsie), adesea descrise ca lumini mici, pâlpâitoare și sclipitoare.
Rata progresiei bolii și gradul de pierdere a vederii variază de la o persoană la alta. Unele cazuri extreme pot evolua rapid în decurs de două decenii, altele un curs lent care nu duce niciodată la orbire completă. Debutul precoce se găsește în forme mai severe de retinită pigmentară, în timp ce pacienții cu afecțiuni mai ușoare (de exemplu, autosomal dominant) pot dezvolta boala în deceniul al cincilea sau al șaselea de viață. În familiile cu retinită pigmentară legată de X, bărbații sunt afectați mai des femeile, pe de altă parte, transmit caracteristica genetică (ele poartă gena modificată pe cromozomul X) și manifestă mai rar simptomele tulburării.
Complicații
Retinita pigmentară va continua să progreseze, deși încet. Cu toate acestea, orbirea completă este rară, dar poate apărea o reducere semnificativă a vederii periferice și centrale.
Pacienții cu retinită pigmentară dezvoltă adesea umflături ale retinei (edem macular) sau cataractă la o vârstă fragedă. Aceste complicații pot fi tratate dacă interferează cu vederea.
Boli conexe
În mod obișnuit, un pacient cu retinită pigmentară nu are alte tulburări și în acest caz vorbim de retinită pigmentară „nesindromică” sau simplă. Cu toate acestea, mai multe sindroame împărtășesc unele simptome clinice cu această boală a ochilor; cel mai frecvent este sindromul Usher, care afectează aproximativ 10-30% din toți pacienții cu retinită pigmentară și este asociat cu pierderea auditivă congenitală sau progresivă. Cu toate acestea, în amauroza congenitală a lui Leber, copiii pot orbi sau aproape orbi în primele șase luni de viață. Alte boli legate de retinita pigmentară includ sindromul Bardet-Biedl și boala Refsum.
Cauze
Boala poate fi cauzată de o serie de defecte genetice: de fapt, există mai multe gene care, dacă sunt afectate de alterare, pot provoca fenotipul retinitei pigmentare. Acestea codifică în mod normal proteinele implicate în cascada de transducție care permite vederea, factorii de transcripție celulară. (care trimit mesaje eronate către celulele retiniene) sau pentru elemente care alcătuiesc structura fotoreceptorilor. Mutațiile genetice moștenite sunt prezente în celule încă din momentul concepției; anomalii comune includ cele ale genelor RP1 (în retinita pigmentară-1, autosomală dominantă) , RHO (RP4, autosomal dominant) și RDS (RP7, autosomal dominant). Cauzele non-ereditare ale retinitei pigmentare sunt rare, dar posibilitatea găsirii unui caz izolat (mutație spontană), în care nu este prezent un istoric familial de boala.












.jpg)